Document Type : Original Article
Authors
1 Department of Organic Chemistry, Faculty of Chemistry, University of Mazandaran, Babolsar, 47416- 95447, Iran
2 Department of Microbiology, Faculty of Science, University of Mazandaran, 47416-95447 Babolsar, Iran
3 Department of Environmental Health Engineering, Faculty of Paramedical Sciences, Babol University of Medicinal Sciences, Babol, Iran
Abstract
We synthesized a series of new azo-based sulfonamides 8a-l via multistep chemical processes including chlorosulfonation, nucleophilic substitution, diazotization, and coupling reactions. The synthesized compounds were characterized using various physical and spectral techniques such as melting point, IR, 1H- and 13C-NMR, Mass, and elemental analysis. We evaluated the antibacterial and anticancer activities of compounds 8a-l. The cytotoxicity of these compounds was assessed on the MCF-7 breast cancer cell line and the MCF-10 human normal cell line after 48 h exposure. Notably, compound 8h demonstrated significantly higher cytotoxicity against MCF-7 (IC50 = 0.21 µM) while showing minimal toxicity towards the MCF-10 human normal cell line. To gain insights into the molecular interactions, we utilized molecular docking to predict the binding affinity of these compounds to the FGFR2 kinase receptor structure (PDB ID: 4J98). Compound 8h exhibited the highest docking score, consistent with our experimental results and demonstrating favorable protein-substrate interactions. In addition, we performed ADME prediction of the compounds, indicating their potential as lead drug candidates. Furthermore, we evaluated the antibacterial activity of compounds 8a-l against Gram-positive and Gram-negative bacteria. Compound 8i showed the strongest antibacterial activity against Staphylococcus aureus, a Gram-positive pathogen. This study provides valuable insights into the biological activities of azo-based sulfonamide derivatives, establishing their potential as both anticancer agents and antibacterial compounds.
Graphical Abstract
)
Keywords
Main Subjects
Introduction
Despite breakthroughs in science and research, cancer remains the second leading cause of death in both industrialized and developing countries. In 2020, there were 19.3 million new cancer cases and almost 10 million deaths reported globally. By 2040, it is estimated that there will be around 29.5 million new cases detected and 16.4 million deaths worldwide. According to the American Cancer Society, there are currently over 227 identified forms of cancer, with breast cancer being the most lethal and the second leading cause of cancer-related death among women [1-4].
Pharmaceutical chemists are required to create innovative anticancer medications that are highly selective, more potent, and have fewer side effects and resistance because radiation therapy, chemotherapy, and surgery have proven to be unsuccessful therapeutic options [5].
A major challenge for medicinal chemists is managing side effects such as osteoporosis,
musculoskeletal pain, and cardiovascular diseases, which can arise from prolonged use of anticancer agents despite their high clinical efficacy in treating breast cancer [6-7].
Given that cancer is a multifaceted disease involving multiple genes, the development of multi-targeted anticancer drugs to improve therapeutic efficacy with reduced toxicity to normal tissue and high effectiveness remains a crucial target in the field of anticancer drug research [8].
On the other hand, sulfonamides are valuable pharmacophores because of their high efficiency, low toxicity, low cost, and wide use in drug research [9]. They have a diverse range of biological actions, including antibacterial [10], anti-inflammatory [11], antifungal [12], HIV protease inhibitors [13], antiviral [14], antimalarial [15], anticancer [16-20], and insulin-releasing [21]. The high biological activity of sulfonamides may be attributed to the specific interaction of the sulfur atoms with enzymes that have sulfhydryl groups in their active sites [22].
Some sulfonamide derivatives have been commercially approved by the FDA as anticancer drugs, such as Amsacrine (1) [23], Pictilisib (2) [24], Pazopanib (3) [25], and Indisulam (4) [26]. Recently, it was reported that sulfonamides containing SO2-morpholine moiety such as compounds (5) [27], (6) [28], and (7) [29], exhibited significant anticancer activity against the MCF-7 cell line (Figure 1).
The presence of an oxygen atom in the morpholine ring can facilitate donor-acceptor interactions with substrates, leading to the creation of stable complexes with target molecules [30-31].
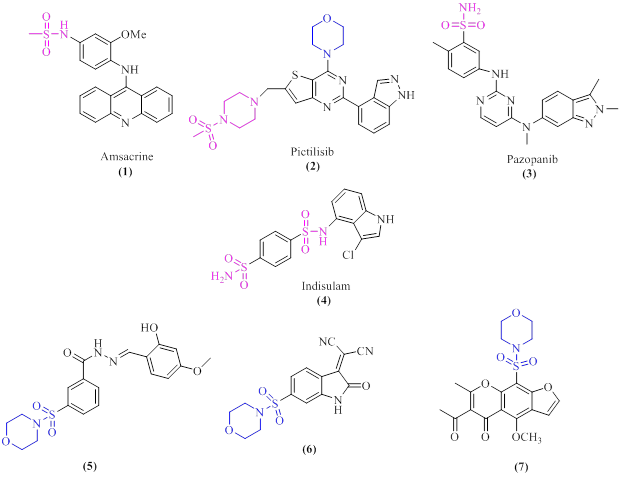
Figure 1: The structure of some anticancer-sulfonamide drugs
In addition, azo compounds have demonstrated effectiveness in a variety of biological activities, including antiviral [32], antibiotic [33], antifungal [34], anticancer [35-37], antioxidant [38], and anti-inflammatory [39]. Some azo compounds have especially displayed cytotoxic effects against the MCF-7 breast cancer cell line [40-42]. It could be due to the azo-linking unit that can form a powerful hydrogen bond with the protein's active site and inhibit the growth of cells [43].
Furthermore, azo-based sulfonamides have shown remarkable anticancer activity. In 2020, N. M. Saleh et al. designed diazepam-bearing sulfonamides with potent anticancer activity against human breast cancer HepG2, HCT-116, and MCF-7 cell lines with IC50 = 8.98 ± 0.1, 7.77 ± 0.1, and 6.99 ± 0.1 μM respectively [44]. In 2023, K. E. Anwer et al. reported new sulfonamides attached to heterocyclic scaffolds such as isoxazole, pyrazole, triazole, and triazine, which were evaluated for growth inhibition of MCF-7 (IC50 = 6.37 μM), HCT-116 (IC50 = 8.44 μM), and HepG2 (IC50 = 6.23 μM) tumors by dual targeting the VEGFR-2 (vascular endothelial growth factor receptor) and EGFR T790M (epidermal growth factor receptor) enzymes [45]. Other recent investigations focused on the anticancer activity of azo-based sulfonamides conjugated with substituted azobenzenes, chromenes, and hydrazones [36,46-48].
In this context, we aimed to create new azo-based sulfonamides and evaluate their cytotoxic potential against the MCF-10 normal cell line and the MCF-7 breast cancer cell line. The results prompted further examinations to reach a deep insight into the mechanism of action of the synthesized compounds and their pharmacokinetic properties via the in vivo ADME (absorption, distribution, metabolism, and excretion) prediction. Molecular docking studies were also conducted to understand the expected binding interactions of the target compounds with FGFR2 (Fibroblast Growth Factor Receptor) active sites and likewise, evaluate their ability to inhibit Gram-positive and Gram-negative bacteria.
Experimental
All starting ingredients and solvents exhibited high purity and were obtained from commercial suppliers such as Sigma-Aldrich and Fluka. The progress of the reactions was monitored by thin-layer chromatography (TLC) on silica gel plates. The chemical structures of the synthesized compounds were confirmed by melting points, infrared (IR) spectroscopy, 1H-nuclear magnetic resonance (NMR) spectroscopy, 13C-NMR spectroscopy, and mass spectrometry. The melting points were determined in open capillary tubes using the Electrothermal 9100 apparatus (Keison Products, Essex, UK). Infrared spectral studies of compounds were performed using a Bruker FT-IR (Bruker, Karlsruhe, Germany) spectrophotometer instrument with the KBr disc method, and spectra were acquired in the 500-4000 cm-1 range. Elemental analysis was carried out by a CHNS-O analyzer (Perkin-Elmer 2400 II).
The 1H- and 13C-NMR spectra were acquired in deuterated dimethyl sulfoxide (DMSO-d6) using a Bruker DRX400 AVANCE (Bruker, Ettlingen, Germany) instrument at (300 and 75 MHz, respectively). For all recorded NMR spectra, the chemical shifts were reported as values in parts per million (ppm) relative to tetramethylsilane. The Finnigan-MAT-8430 mass spectrometer (Agilent 5975C, Wilmington, DE) was used to produce high-resolution mass spectra (70 eV).
Synthesis of 4-Acetamidobenzen Sulfonyl Chloride 2
A mixture of chlorosulfonic acid (100 mmol, 7 mL) and acetanilide 1 (22 mmol, 3 g) was stirred at 0-5 °C for 30 min, and then the mixture was heated at 80 °C for 60 min and allowed to cool to room temperature. Ice water was added to the reaction mixture to precipitate 4-acetamidobenzenesulfonyl chloride 2, which was then collected by filtration. Yield: 90%, Mp: 145 °C.
Synthesis of Sulfonamide Derivatives 5a-d
4-acetamidobenzenesulfonyl chloride 2 (20 mmol, 5 g) was mixed with (8 mmol) of appropriate amines 3a-d in pyridine (65 mmol, 5 mL) at 0-5 °C for 2 h, and the reaction mixture was agitated for another 3 h at room temperature, water was added, and then it was filtered and dried to obtain intermediates 4a-d. An aqueous solution of NaOH (5 M, 15 mL) was mixed with 10 mL of methanol and intermediates 4a-d and the reaction mixture was heated at reflux condition for 3 h. The reaction's progress was monitored by TLC. Following the addition of HCl (2 M) solution, the reaction mixture was adjusted to a pH of 6 and stirred until the product was formed, and then it was recrystallized from ethanol/water to give pure products 5a-d.
General Technique for the Synthesis of Azo-Based Sulfonamides 8a-l
Sulfonamides 5a-d (5 mmol) were dissolved in dilute hydrochloric acid (2.5 mL HCl in 10 mL of H2O) and cooled to 0-5 °C in an ice bath while being stirred. A cold aqueous sodium nitrite solution (7 mmol, 0.5 g, 5 mL H2O) was added dropwise to the above solution, and the mixture was rapidly stirred for about 1 h to afford diazonium salt solution 6a-d.
Compounds 7a-e (5 mmol) were dissolved in an aqueous solution of K2CO3 (7 mmol, 1 g, in 15 mL H2O), then maintained in an ice bath at 0-5 °C with stirring. The obtained 6a-d diazonium salt solution was added dropwise to the stirred alkaline solution over 30 min, and the pH was kept at 6, the resultant reaction mixture was stirred for 2 h and maintained at 0-5 °C while the progress of the reaction was monitoring by TLC. The crude product 8a-l was filtered off, washed several times with water, dried, and recrystallized with ethanol.
(E)-4-((4-Hydroxy-6-methyl-2-oxo-2H-pyran-3-yl) diazenyl) Benzenesulfonamide (8a)
Light orange powder; Yield 87%; m.p. 297-300 °C. IR (KBr, cm-1): 3359, 3257 (OH, NH2), 3100 (aromatic CH), 1737 (C = O), 1645 and 1585 (C = C), 1513 (N=N), 1333 (SO2, asym), and 1149 (SO2, sym). 1H-NMR (300.1 MHz, DMSO-d6): δH 2.23 (s, 3H, CH3), 6.08 (s, 1H, =C-H), 7.46 (s, 2H, NH2), 7.83 (d, 2H, 3JHH = 8.7 Hz, Ar-H), 7.93 (d, 2H, 3JHH = 8.7 Hz, Ar-H), and 15.55 (s, 1H, OH). 13C-NMR (100 MHz, DMSO-d6): δC 20.32, 107.87, 118.13, 123.75, 127.96, 142.42, 143.80, 159.03, 167.80, 180.96. MS (m/z, %): 309 (M+, 99), 280 (14), 197 (19), 153 (100), and 125 (60). Elemental analysis calculated for C12H11N3O5S: C 46.60; H 3.58; N 13.59; and S 10.37 %. Found: C 46.46; H 3.57; N 13.55; and S 10.41 %.
In Vitro Anticancer Studies
The cell viability assay used in vitro MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay to investigate the effect of azo-based sulfonamides on the MCF-7 (a human breast cancer cell line) and MCF-10 (a human mammary epithelial cell line) [49-50]. In brief, the MCF-7 and MCF-10 cells were grown and seeded at a density of (2 × 104) in a 96-well plate. The medium was incubated at 37 °C for 48 and 72 hours with concentrations of the target azo-based sulfonamides (15, 31, 62, 125, and 250 µM) in DMSO. The supernatant was then poured out and rinsed once with PBS. After being replaced with (0.5 mg/mL) of MTT in each well, the cells were incubated for 4 hours at 37 °C. The formazan purple crystals were dissolved in 100 mL of DMSO and the absorbance at 570 nm was measured with a microplate reader. The percentage cytotoxicity was calculated using Equation (1) for all the tested samples.
% Cytotoxicity = 1- OD test compound /OD control (1)
The concentration required to diminish cell viability by 50% compared to maximal (control) viability (IC50) was used to illustrate the results. The experiment was performed three times to test the cytotoxicity of the compounds, and the mean of the data (± SD) was determined by the GraphPad Prism 9 software.
Docking Study Assay
Molecular docking is a valuable approach for creating pharmacological compounds that effectively target specific proteins and nucleic acids by considering various non-covalent interactions between these molecules [51-53].
The X-ray crystal structure of FGFR2 (PDB-ID 4J98) was selected from the RCSB Protein Data Bank (www.rcsb.org). The ligands were ready for docking through LigPrep, and the design panel sketched them in 3D.
The Schrodinger Protein Preparation Wizard was performed to delete water molecules and crystallographic agents; amino acid protonation states for pH 7.0 were defined; and the hydrogen bonding network was optimized. Grids for molecular docking were created using the co-crystallized-bound ligand.
In the docking protocol, the co-crystallized ligands were subjected to re-docked simulations, and the root-mean-square deviation (RMSD) between the original and re-docked ligands was calculated. The RMSD values were used to evaluate the consistency of the docked ligand positions in the protein target pocket.
In Silico ADME Assay
Today, it is possible to predict the pharmacokinetic characteristics, biological properties, and pharmaceutical similarity of products before starting preclinical research by performing computational methods [54-55].
The drug’s likeness features of azo-based sulfonamides 8a-l were utilized to determine the drug-like action. In this study, the drug-likeness of the synthesized compounds was investigated through the SwissADME program and compared their similarities and superiorities with the standard drug.
When 2D structures were transformed to canonical SMILES format, the ADME properties of the suggested chemical compounds were calculated using the free online SwissADME tool (http://www. swissadme.ch/index.php).
Agar Well Diffusion Test
Using the disc diffusion method, all of the generated compounds 8a-l were evaluated for antibacterial activity against four bacterial strains, two Gram-positive bacteria (Staph. aureus and Bacillus subtilis) and two Gram-negative bacteria (Escherichia coli and Pseudomonas aeruginosa) [56-59].
100 μL of the bacterial suspension was uniformly impregnated onto LB agar plates. Each of the targets was dissolved in dimethyl sulfoxide in 20 μg mL-1 concentration separately, absorbed into sterile paper discs, and incubated at 37 °C in an incubator for 24 h. The negative control used was dimethyl sulfoxide, Gentamicin (10 µg/disc), and Chloramphenicol (30 µg/disc) concentrations were employed as the positive controls for the microorganisms.
Results and Discussion
Chemistry
The synthetic pathway for producing the desired azo-based sulfonamides 8a-l is depicted in Scheme 1. The synthesis of sulfonamide derivatives 5a-d involved three distinct steps, as reported in the literature [60]. With slight modification. Under solvent-free circumstances, acetanilide 1 and chlorosulfonic acid were mixed and stirred at 80 °C to form 4-acetamidobenzensulfonyl chloride 2 in high yield (90%). Subsequently, compound 2 reacted with amines such as ammonia 3a, primary amines (3b and 3c), and morpholine 3d to afford compounds 4a-d. Hydrolysis of compounds 4a-d in alkaline methanolic aqueous solution produced compounds 4-amino-N-substituted benzenesulfonamides 5a-d, which then diazotized to diazonium salts 6a-d using sodium nitrite and HCl (pH=3.5). Finally, compounds 6a-d were coupled with electron-rich compounds including triacetic acid lactone 7a, 4-hydroxy-6-methyl-2-pyrone 4-hydroxycoumarin 7b, salicylaldehyde 7c, vanillin 7d, and 3,5-dimethoxyphenol 7e in the presence of K2CO3, which led to azo-based sulfonamide products 8a-l in good to high yields (75-90%) (Table 1).
The structure of synthesized compounds 8a-l was characterized using their IR, MS, 1H-, and 13C-NMR spectral data. As an example, the identification of compound 8a is explained. The IR spectrum of compound 8a revealed an absorption band at 3359 and 3257 cm-1 indicating the presence of the OH and NH2 groups, respectively. Other characteristic signals appeared at 1737 cm-1 (C=O, stretching), 1513 cm-1 (N=N, stretching), and two absorption bands at 1333 and 1149 cm-1 (SO2, asymmetric and symmetric stretching). The 1H-NMR spectrum of compound 8a in DMSO indicated a singlet at 2.23 ppm for the CH3 group, a singlet at 6.08 ppm for the olefinic proton of pyran 2-one ring, and a singlet at 7.46 ppm for the NH2 group. Also, two doublets appeared at 7.83 and 7.93 ppm (3J = 8.7 Hz) for the phenyl protons and a signal at 15.55 ppm for the OH proton. The 13C-NMR spectrum of 8a revealed 10 signals in appropriate ppm consistent with the postulated structure. At 20.3 ppm, the methyl group was discovered, and at 180.9 ppm, the C=O group was detected. The remaining signals at 107.8, 118.1, 123.7, 127.9, 142.4, 143.8, 159.0, and 167.8 ppm are also associated with the aromatic carbons of 8a.
The mass spectrum of compound 8a displayed its molecular ion (M+) and base peak at m/z = 309 and 153, respectively. The other peaks at 280, 197, and 125 are associated with [M- (CO+H)], [M-(CH3+ OH+ SO2NH2)], and [M-N2C6H4SO2NH2] ion fragments.The spectra of the remaining compounds 8b-l have been analyzed and are shown in the supplementary material file.
Cytotoxic Screening
The cytotoxic activity of all the target compounds 8a-l was evaluated against the human breast cancer cell line MCF-7 using an MTT assay, and compared with the normal cell line MCF-10 at 48 h and 72 h. The half-maximum inhibitory concentrations (IC50) were computed at various concentrations of 8a-l and the obtained results are presented in Tables 2 and 3. Doxorubicin was used as a chemotherapeutic anticancer drug and, as a positive standard, for the MTT assay investigation. A comparison of the data in Table 2 with those in Table 3 exhibits that the maximum cytotoxic effects of compounds 8a-l are observed after 48 h and slightly decreased after 72 h. Therefore, the cytotoxicity of the tested compounds was investigated after 48 h (Table 2).
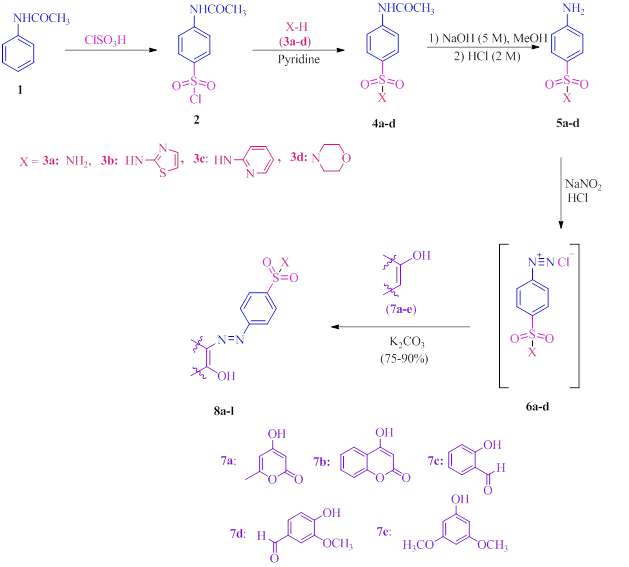
Scheme 1: A general strategy for the synthesis of azo-based sulfonamides 8a-l

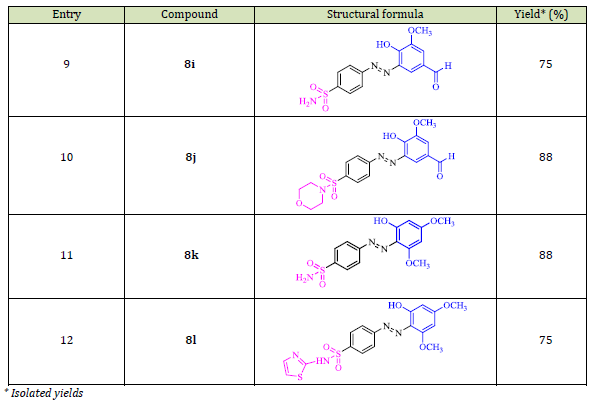
As shown in Table 2 compounds 8a, 8b, 8e, and 8g-k exhibited higher inhibition activity (IC50 = 0.48±0.008, 2.81±0.007, 2.82±0.01, 1.41±0.013, 0.21±0.008, 0.18±0.008, and 0.19±0.006, and 2.38±0.009 μM, respectively) than doxorubicin (IC50 = 3.42 μM) after 48 h exposure. Among them, compounds 8h-j displayed the highest cytotoxic activity against the MCF-7 cancer cell line, with IC50 values of 0.18±0.008, 0.19±0.006, and 0.21±0.008 μM, respectively. The presence of SO2-NH2 and SO2-morpholine systems especially the carbonyl group in derivatives 8h-j enhanced the antitumor activity and led to results better than the reference drug [61-62]. Since compound 8h showed the lowest cytotoxic effect against the normal cell line (IC50 = 75.01±0.006 μM), it is considered the most effective compound among all compounds in this study.


The effectiveness of the compound 8a-l concentrations on the cytotoxicity against the MCF-7 cell line in 48h was studied and depicted in Figure 2. It is obvious in this Figure that the cytotoxic activities of all compounds increased from 15 to 250 μM, with the highest activity at 250 μM concentration.
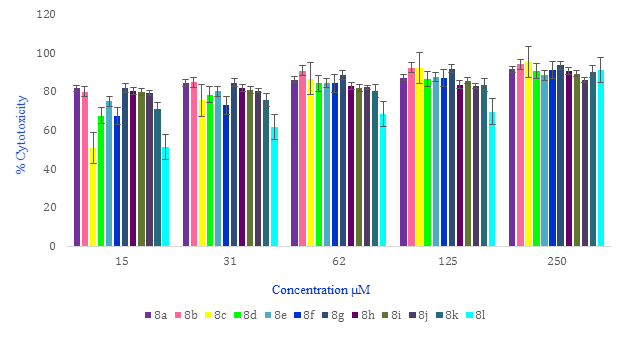
Figure 2: Concentration-dependent cytotoxic potential of compounds against the MCF-7 cell line ranging from 15 to 250 μM in 48 h. Mean ± S.D is indicated by the error bar
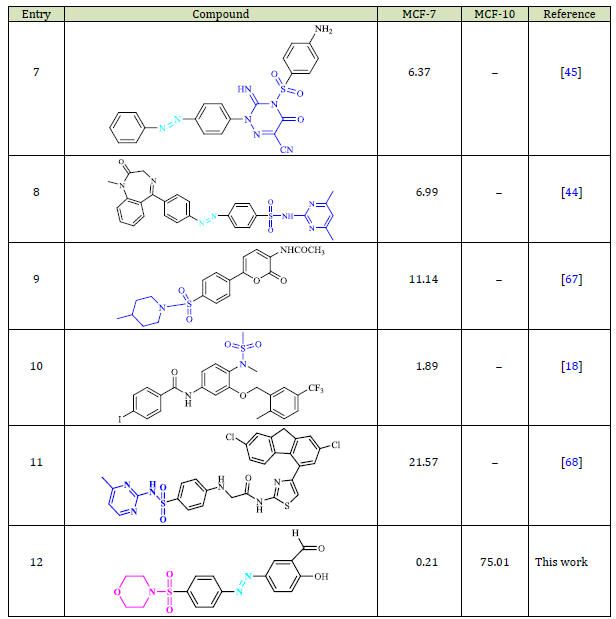
Comparison of Cytotoxic Activity of Compound 8h with the Other Reported Sulfonamides
The cytotoxic activity of compound 8h has been compared with those reported in the literature in Table 4. As seen, compound 8h is more active than the other sulfonamides (entry 12 compared to entries 9-11) and azo-based sulfonamides (entry 12 compared to entries 7 and 8). In addition, compound 8h displayed higher cytotoxic activity compared with compounds in entries 5 and 6 that possess sulfonamide (-SO2NH-) and morpholine moieties in their structure. Furthermore, compound 8h exhibited higher cytotoxicity compared with compounds in entries 1, 2, and 4 containing sulfonamide-morpholine moiety. Although the cytotoxic effect of the compound in entry 3 is higher than that for compound 8h, it was not tested against the MCF-10 cell line. The higher activity of 8h could be due to the formation of more hydrogen bonds than the other compounds in Table 4, which is consistent with the obtained results from the molecular docking study.

ADME Profile Study
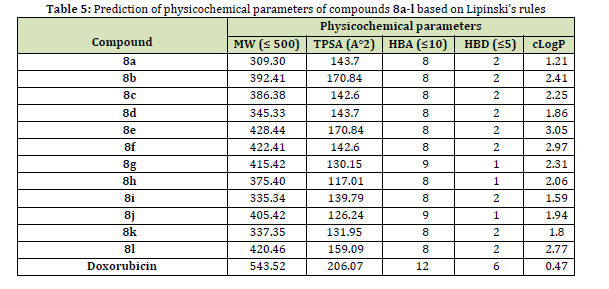
The drug-likeness of a molecule is a measure of how likely it is to be successfully developed into an oral drug. This assessment is based on the structure and physicochemical properties of the molecule [69]. Five of Lipinski’s rules including molecular weight (MW), the topological polar surface area (TPSA), the number of hydrogen bond donors (HBD), the number of HB acceptors (HBA), and lipophilicity (cLog P), are good approaches to predicting the drug-like properties of compounds.
The physicochemical parameters for the synthesized azo-based sulfonamides 8a-l were calculated by SwissADME software, as shown in Table 5. All the physicochemical parameters of compounds 8a-l in Table 5 are within an allowable extent, indicating that these compounds are capable of being considered as druggable compounds. In addition, the bioavailability score and Lipinski’s violation were calculated to be 0.55 and zero, respectively. These values confirmed further that the compounds 8a-l are potentially drug-likeness compounds.
In Silico Studies Molecular Docking
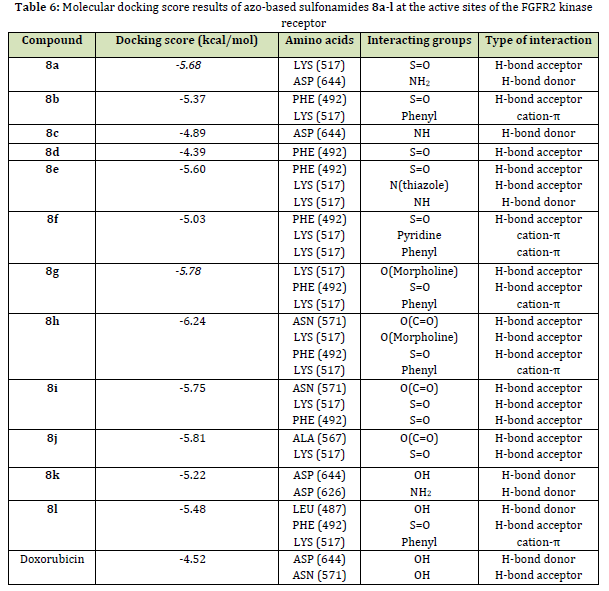
FGFR2 kinase receptor (Homo sapiens) is a receptor tyrosine kinase expressed on the cell membrane that plays an important role in developmental and adult cells [70]. Dysregulation of FGER2 is involved in various types of cancers such as breast cancer [71-72]. In this work, the molecular docking study of the FGFR2 kinase receptor (PDB code: 4J98) with the synthesized compounds 8a-l was performed by Schrodinger software. Among synthesized compounds, 8h exhibited the highest docking score (-6.24 kcal/mol) and strong binding interaction with the backbone of the FGFR2 kinase receptor, as indicated in Table 6. Likewise, Figure 3 (a and b) exhibits the formation of three hydrogen bonds between the oxygen atom of the sulfonamide group with PHE 492, the oxygen atom of morpholine with LYS 517, and the oxygen atom of the aldehyde group with ASN 571. In addition, there is a cation-π interaction between the phenyl group of 8h with LYS 517. These results are consistent with the obtained results from the MTT assay investigation. However, most of the investigated compounds exhibited a high affinity toward the target proteins. This is further confirmed by comparing their ducking scores with that of reference anticancer drug (Table 6).
Accelerated development is warranted for the compound 8h, which exhibits promise as a candidate for providing breast cancer patients with more durable and effective disease control. This development signifies a significant turning point in the pursuit of precision medications capable of surmounting the obstacles posed by drug resistance. The findings indicate that the azo-based sulfonamide scaffold exhibits considerable potential as a framework for the development of FGFR2 inhibitors. Particularly, the selectivity and potency of compound 8h against FGFR2 kinase validate this target for the treatment of breast cancer. The fact that it inhibits tumor growth while causing minimal toxicity indicates that targeting FGFR2 may create a favorable therapeutic window. A constraint of the study was that the in vitro assays were exclusive to the MCF-7 breast cancer cell line. Although widely employed, these models fail to comprehensively represent the heterogeneity that characterizes breast cancer. Incorporating a wider range of breast cancer cell lines exhibiting diverse FGFR2 expression levels and mutation statuses into the evaluation would contribute to the reinforcement of the conclusions concerning the efficacy and target validation of compound 8h.
a 
b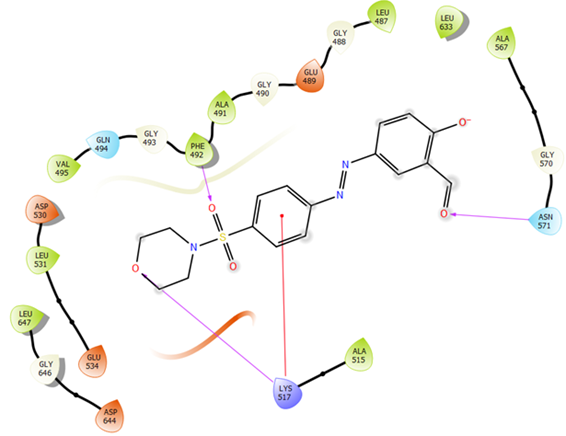
Figure 3: (a) Binding mode of highest-docking-score for compound 8h in the FGFR2 kinase receptor (PDB ID: 4J98) active site (b) 2D interaction of compound 8h with the FGFR2 kinase receptor
Furthermore, the insights gained from the molecular docking simulations regarding the potential binding mode of compound 8h with the FGFR2 kinase domain were extremely valuable. Nevertheless, inherent uncertainties persist due to the absence of an experimental determination of the binding pose. X-ray crystallography analyses of compound 8h in complex with FGFR2 would provide additional support for the computational model's predicted binding mode. Future work will aim to address these limitations by expanding the in vitro testing to additional breast cancer cell lines, conducting X-ray crystallography, performing in vivo efficacy and toxicity studies, and carrying out comprehensive pharmacokinetic profiling.
Antibacterial Activity
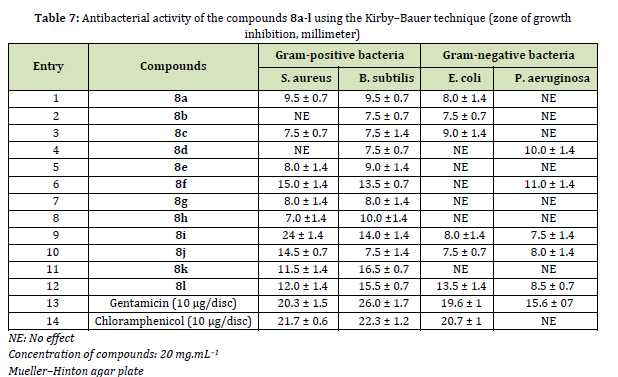
The antibacterial activity of synthesized azo-based sulfonamides 8a-l was assessed against two Gram-positive human pathogenic organisms, Bacillus subtilis (B. subtilis), Staphylococcus aureus (S. aureus), and two Gram-negative bacteria, Pseudomonas aeruginosa (P. aeruginosa), and Escherichia coli (E. coli), using the agar-well diffusion method. Gentamicin and Chloramphenicol were used as positive controls. As indicated in Table 7, compounds 8a-l are more active against Gram-positive bacteria than Gram-negative bacteria which may be attributed to their peptidoglycan outer layer. The thick peptidoglycan layer of Gram-positive bacteria causes them to be easier to absorb antibiotics to kill, as has been reported previously [73]. Among them, 8i has shown the best antibacterial activity against S. aureus (24 ± 1.4 mg/mL).
Conclusion
In this study, we prepared new azo-based sulfonamides 8a-l through chemical processes that included sulfonation, nucleophilic substitution (SN2), deacetylation, diazotization, and finally azo coupling. Short reaction times, mild conditions, and high yields are the noticeable features of this protocol. The cytotoxicity of the synthesized compounds was assessed on the breast cancer cell line MCF-7 and the human normal cell line MCF-10 after 48 h exposure. Compound 8a, 8b, 8e, and 8g-k displayed outstanding activity against the MCF-7 cell line with IC50 values of 0.18 to 2.82 µM, indicating a comparable inhibitory activity to doxorubicin (IC50 = 3.42 µM). Furthermore, compound 8i showed the strongest antibacterial action against Staphylococcus aureus, a Gram-positive pathogen with a value of 24 μg/mL.
The molecular docking study of the compounds 8a-l against the FGFR2 kinase receptor (PDB ID: 4J98) indicated that compound 8h had the highest docking score (-6.24 kcal/mol).
The carbonyl oxygen and sulfur moieties in the compounds 8h, 8i, and 8j interact with essential amino acid residues to form hydrogen bonds, contributing to their inhibitory activity against FGFR2. Interestingly, the ADME parameters prediction indicated that most of the synthesized compounds have acceptable pharmacokinetics.
Based on the results, compound 8h exhibits potential as a viable candidate for inhibiting the growth of breast cancer cell MCF-7 (IC50 = 0.21 µM) and with low toxicity to the normal cell line MCF-10. The comparison of the cytotoxic activity of compound 8h with other sulfonamides and azo-based sulfonamides reported in the literature highlights its superior efficacy, indicating a novel finding in the field.
Its accelerated development could provide breast cancer patients with more effective and durable disease control, addressing the challenge of drug resistance. The azo-based sulfonamide scaffold, particularly compound 8h, shows promise as an FGFR2 inhibitor for breast cancer treatment, validated by its selectivity and potency. However, the study's limitations include the reliance on a single breast cancer cell line, MCF-7, which does not fully represent the diversity of breast tumors. Molecular docking simulations provided insights into the binding mechanism of compound 8h, but experimental determination through X-ray crystallography is necessary for confirmation. Future investigations should expand in vitro testing, utilize X-ray crystallography, conduct in vivo efficacy and toxicity assessments, and establish comprehensive pharmacokinetic profiles. Doing so will help advance the preclinical development of compound 8h as a promising new targeted therapy for FGFR2-driven breast cancers.
Acknowledgments
The authors are thankful to the University of Mazandaran for partial support of this project.
ORCID
Olia Rezaeianzadeh
https://orcid.org/0009-0008-9436-7845
Sakineh Asghari
https://orcid.org/0000-0002-6608-9525
Mahmood Tajbakhsh
https://orcid.org/0000-0002-4856-1937
Mojtaba Mohseni
https://orcid.org/0000-0002-5709-6600
Asieh Khalilpour
https://orcid.org/0000-0002-2401-4306
HOW TO CITE THIS ARTICLE
Rezaeianzadeh, S. Asghari, M. Tajbakhsh, M. Mohseni, A. Khalilpour. Synthesis, Molecular Docking, and Anticancer Evaluation of New Azo-Based Sulfonamides against MCF‑7 Human Breast Cancer Cell Line. Chem. Methodol., 2024, 8(5) 329-350
OPEN ACCESS
©2024 The author(s). This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit: http://creativecommons.org/licenses/by/4.0/
PUBLISHER NOTE
Sami Publishing Company remains neutral concerning jurisdictional claims in published maps and institutional affiliations.
CURRENT PUBLISHER
)